Clinical characteristics.
Bietti’s crystalline dystrophy (BCD, also known as Bietti crystalline corneoretinal dystrophy, or Bietti’s crystalline retinopathy) is a chorioretinal degeneration characterized by the presence of yellow or refractile crystals and/or complex lipid deposits in the retina and (in some patients and to a variable degree) the cornea. Progressive atrophy and degeneration of the retinal pigment epithelium (RPE) / choroid lead to symptoms similar to those of other forms of retinal degeneration that fall under the category of retinitis pigmentosa (RP) and allied disorders, namely: reduced visual acuity, poor night vision, abnormal retinal electrophysiology, visual field loss, and often impaired color vision.
Diagnosis of BCD can be made through clinical findings and genetic testing. BCD may be misdiagnosed because its symptoms are similar to those of other eye disorders that progressively damage the retina (See BCD Misdiagnosis). Identification of biallelic mutations in the CYP4V2 gene by molecular genetic testing can confirm the diagnosis if clinical features are inconclusive.
Suggestive Clinical Findings: 1 2 3
BCD should be suspected in individuals with the following clinical, electrophysiology, and imaging findings.
Symptoms:
Progressive vision impairment:
- night blindness (Nyctalopia),
- distorted vision,
- sensitive to strong light (Photophobia),
- reduced visual acuity,
- visual field loss affecting peripheral and/or central vision.
- reduced contrast sensitivity,
- impaired color vision, and
- eventually legal blindness.
Age of onset, presenting symptoms, and disease severity vary among patients. Vision impairment is progressive. Marked asymmetry between eyes is common.
Who are Affected:
- Gender: BCD affect both men and women
- Onset Age: People with Bietti’s Crystalline Dystrophy (BCD) typically begin noticing vision problems during the second to third decade of life, but the onset ranges from the early teenage years to beyond the third decade.
- Ethnicity: Patients of different ethnic origins have been reported throughout the world, including African, Asian, European or the Middle Eastern descent.
Inheritance pattern and family history:
- BCD is caused by biallelic mutations in the CYP4V2 gene and is inherited in an autosomal recessive pattern. For most BCD patients, their parents are carriers of CYP4V2 mutation but are not affected. BCD can affect both men and women.
- Statistically, BCD incidence rate among the children of carriers of CYP4V2 mutation is 25%. Hence, many BCD patients do not have a family history. Actual incidence rate varies from family to family.
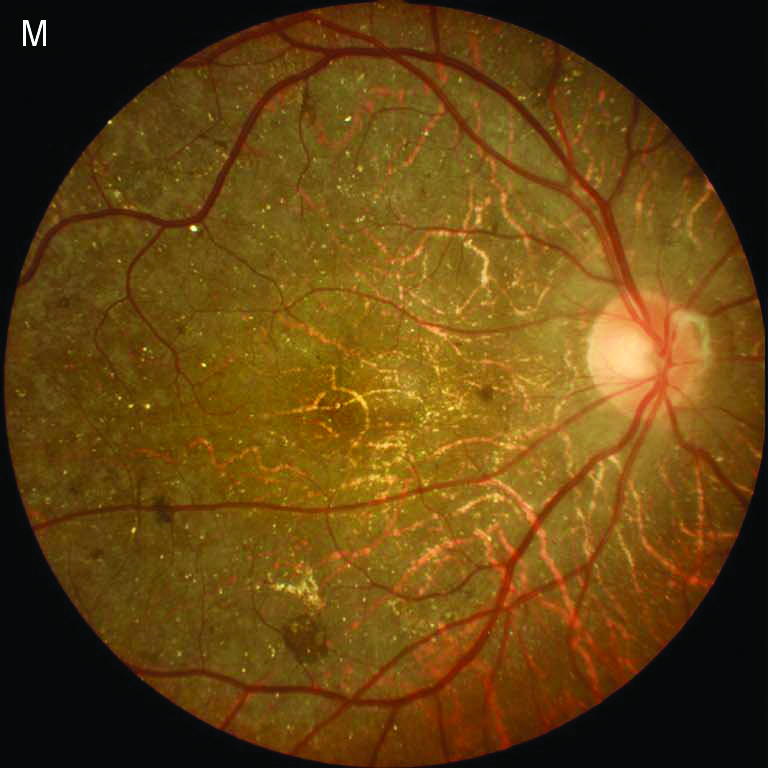
Retina
- Numerous small, glistening yellow-white or refractile crystals scattered throughout the posterior pole may extend to the midperiphery.
- Atrophy of the retinal pigment epithelium (RPE) and choriocapillaris
- Pigment clumping
- Sclerosis of the choroidal vessels
- Patchy hypofluorescent areas of RPE, choriocapillaris atrophy, and a generalized disturbance of the RPE seen on fluorescein angiography
Factsheet on Bietti Crystalline Dystrophy (BCD), a blinding disease
(Click on images to see full image)
| Clinical Phenotype: | Stage 1 | Stage 2 | Stage 3 |
|---|---|---|---|
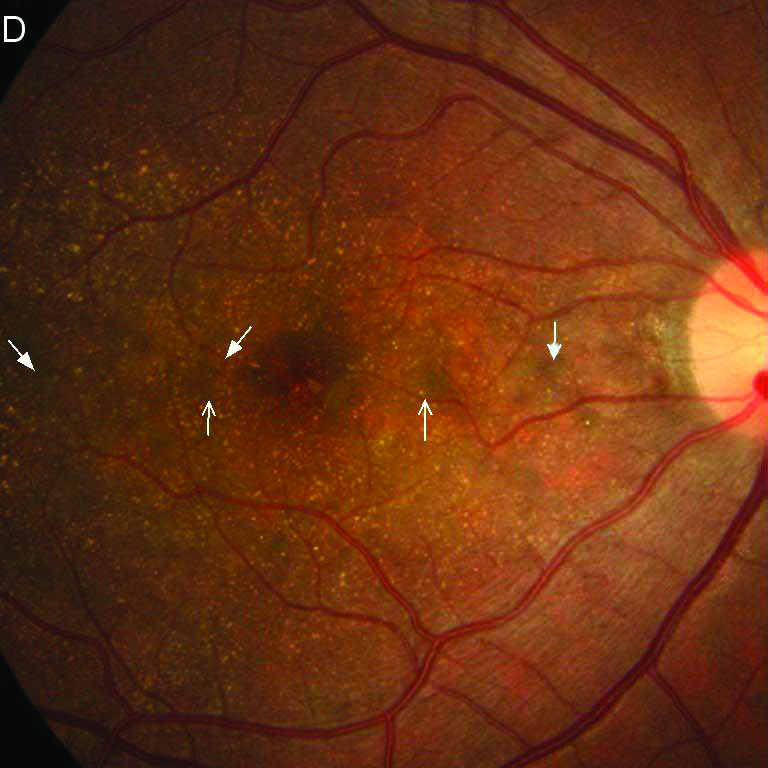
Fundus Photography |
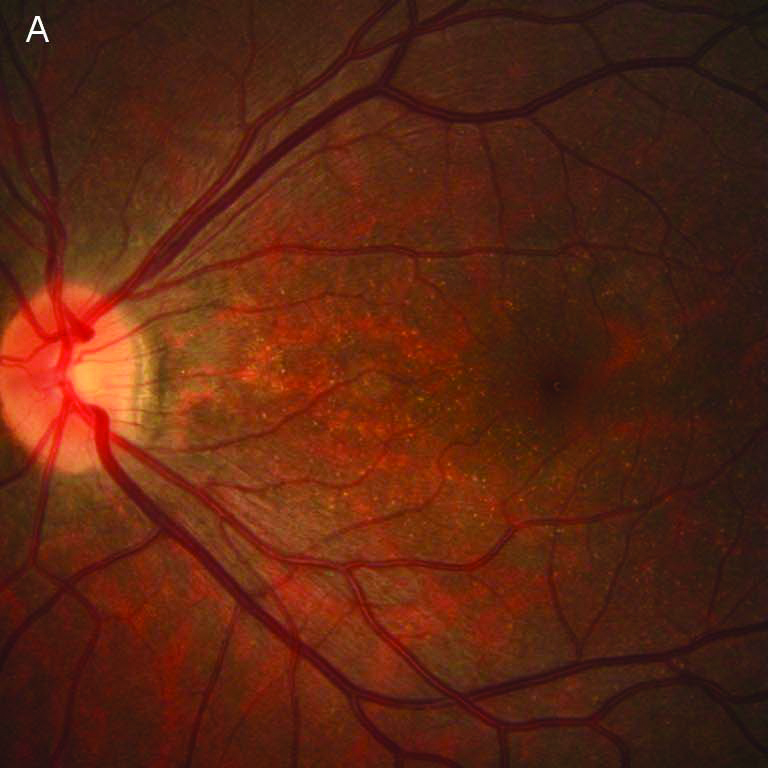 
|
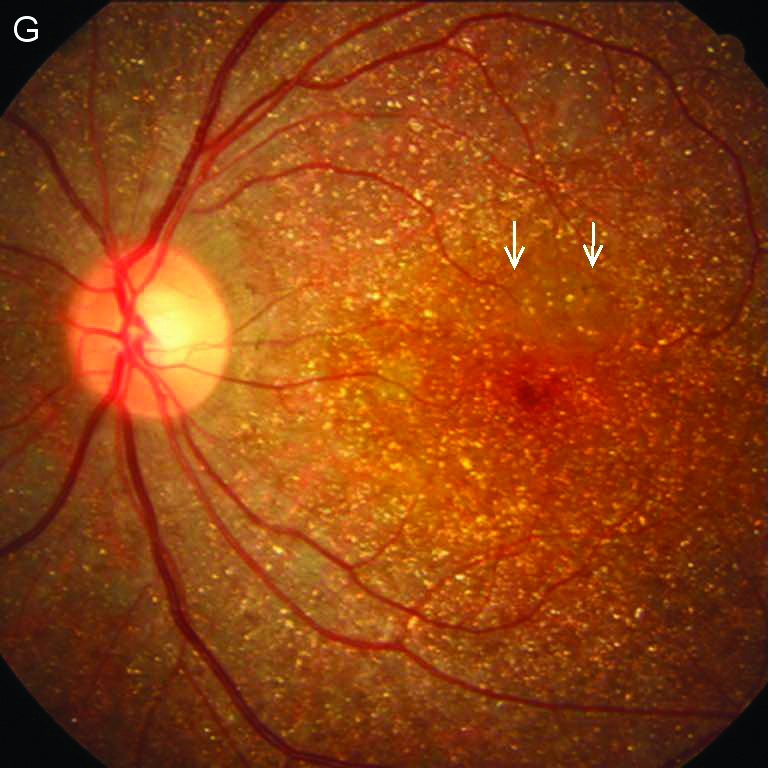 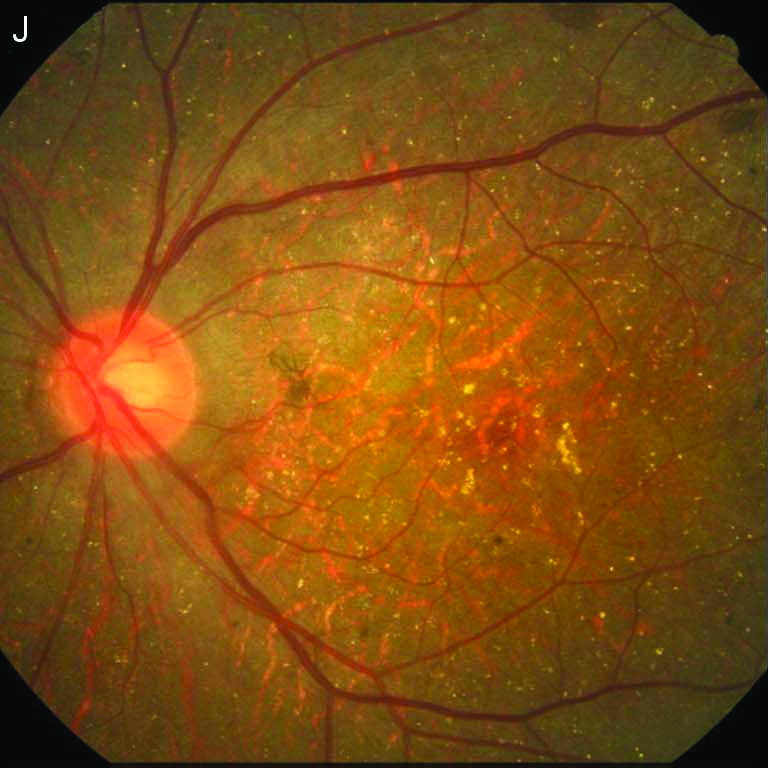
|
|
|
RPE (retinal pigment epithelium) atrophy in the posterior pole.
|
RPE atrophy extends beyond the posterior pole, with RPE-choriocapillaris atrophy appearing markedly in the posterior pole.
|
Prominent diffuse atrophy of the RPE-choriocapillaris complex.
|
|
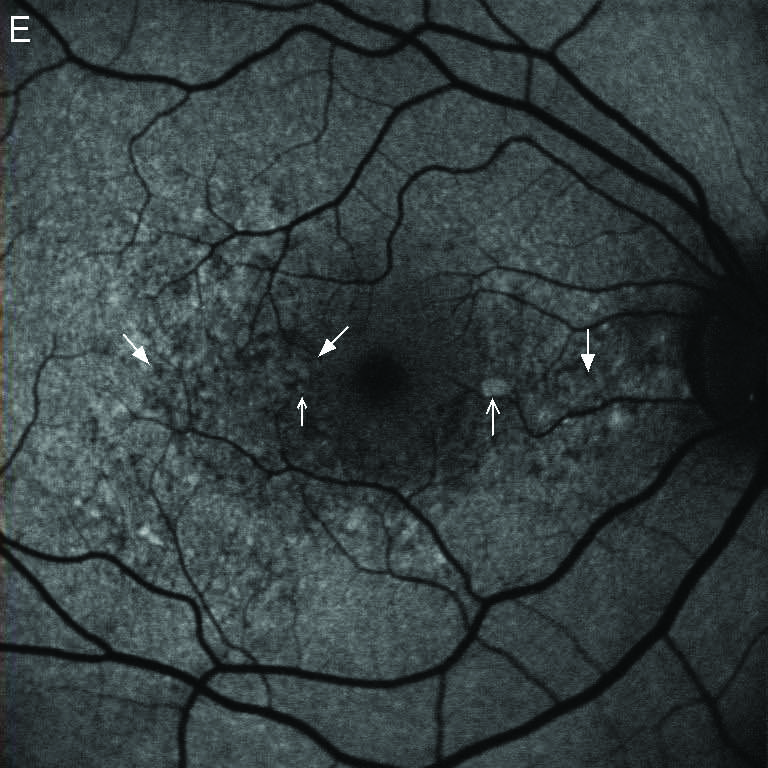
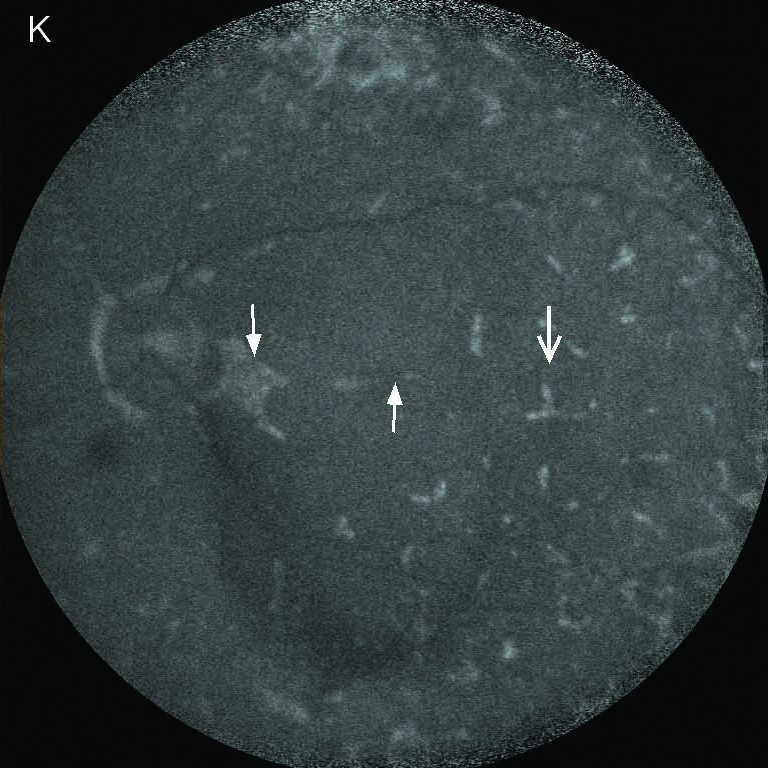
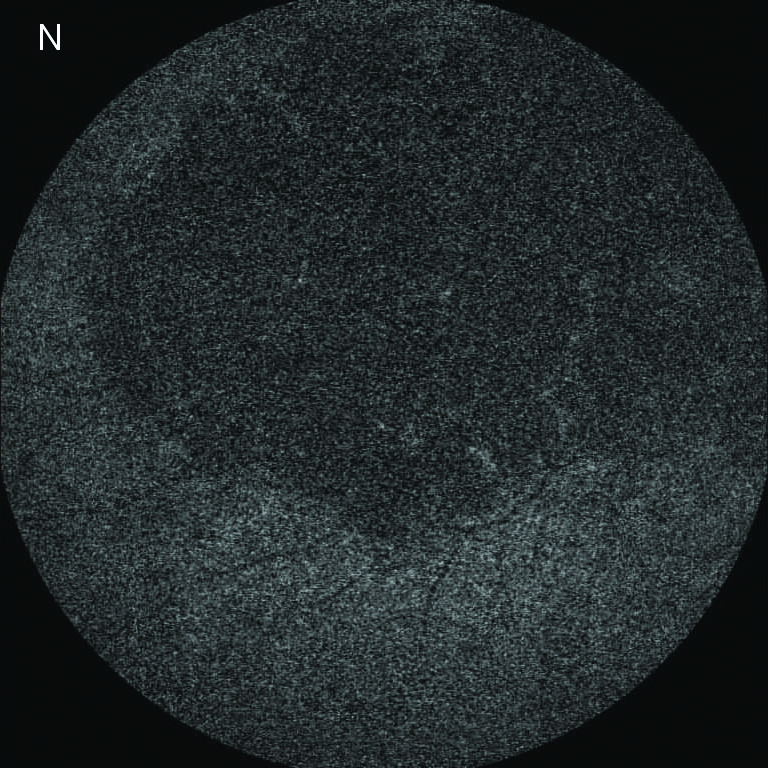
FAF(Fundus Autofluorescence) |
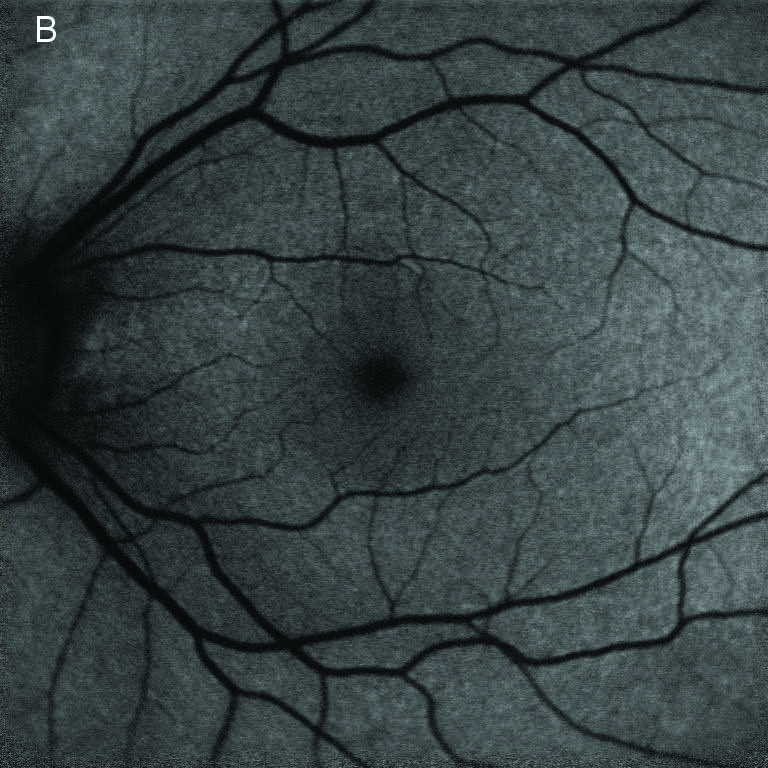 
|
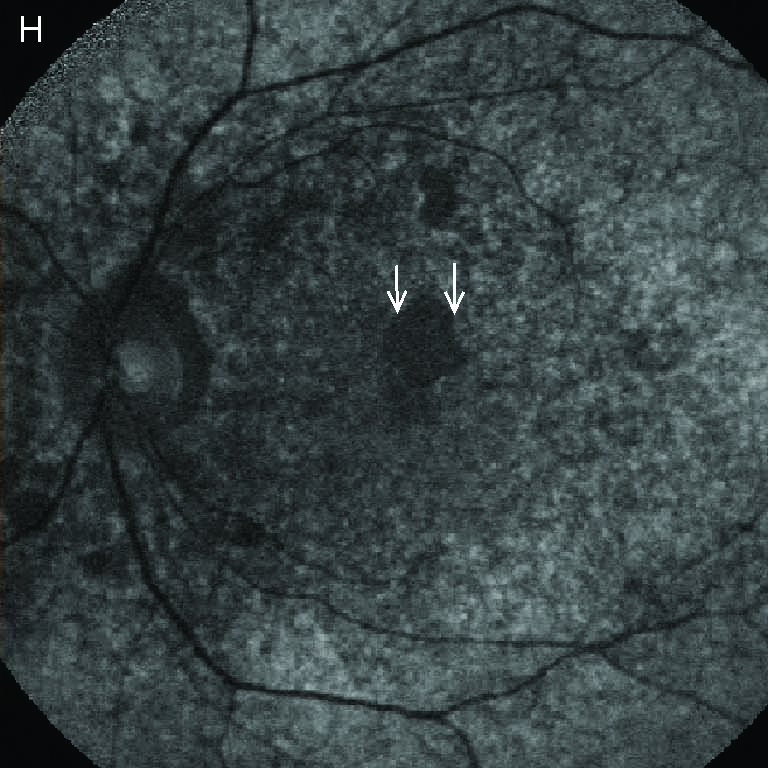 
|
|
|
Normal AF (autofluorescence) in extremely early stage or hyper/hypo-AF confined in the posterior pole.
|
Confluent absent-AF patches and hypo-AF extend beyond the posterior pole.
|
Prominent absent-AF in the posterior pole.
|
|
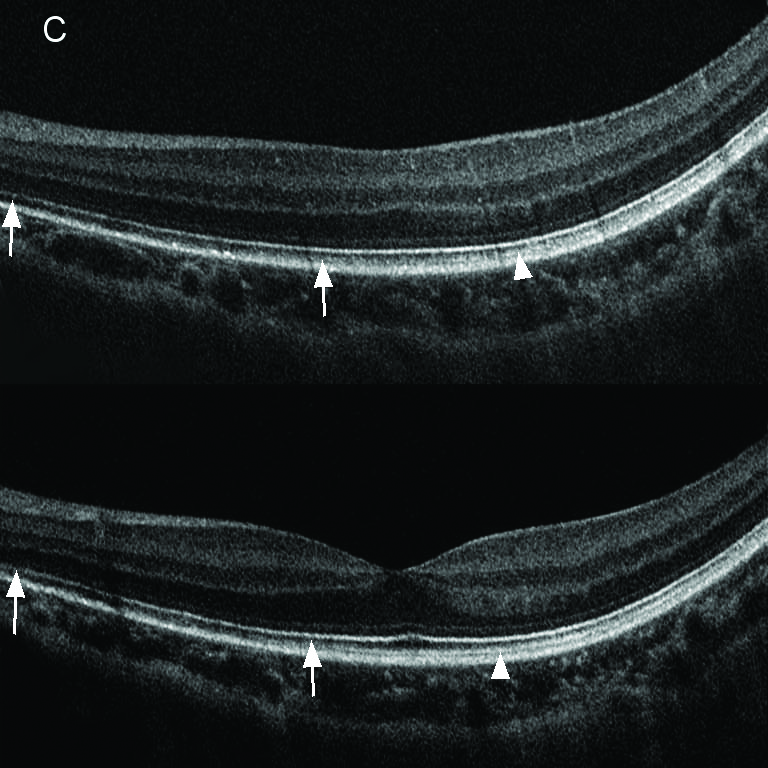
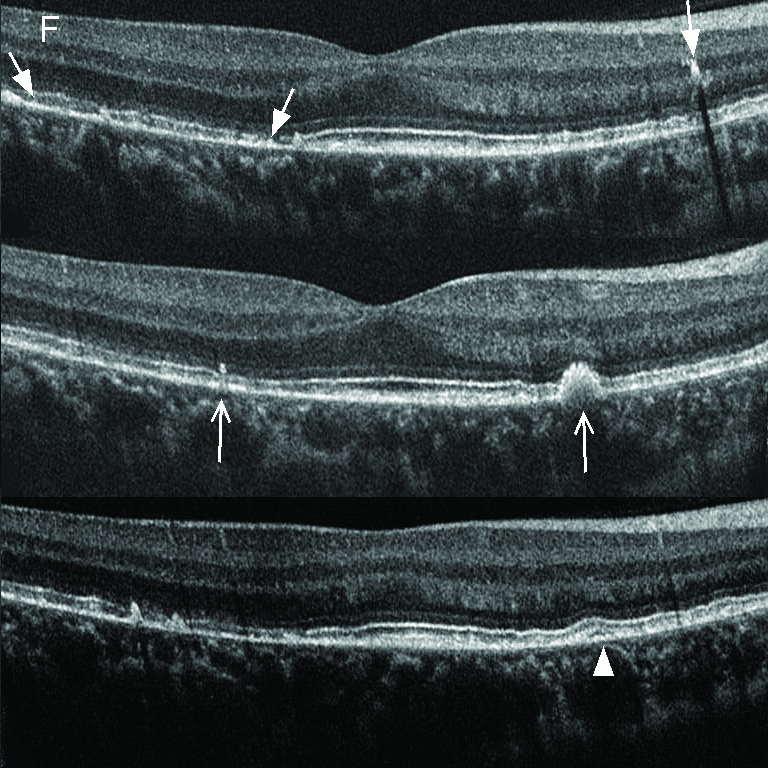
SD-OCT (EDI Mode)(spectral-domain optical coherence tomography) |
|
|
|
|
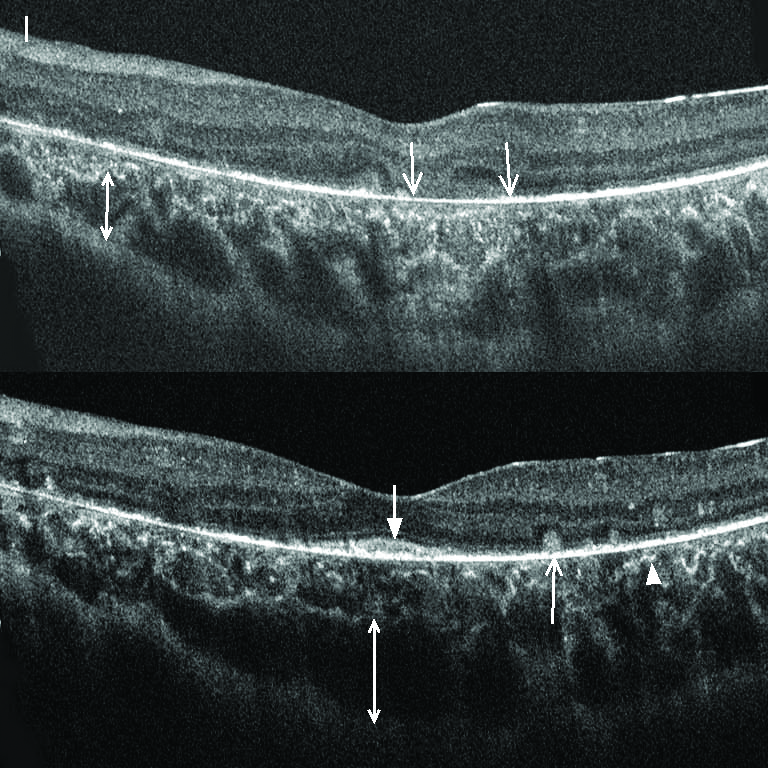
SD-OCT
|
Drusen-like structure, loss of IZ (interdigitation zone) and localized disruption of EZ (ellipsoid zone) in macula.
|
Extensive loss of the outer retina and RPE, with focal residual RPE and/or EZ in macula.
|
Complete loss of RPE and severe dystrophy of the macula.
|
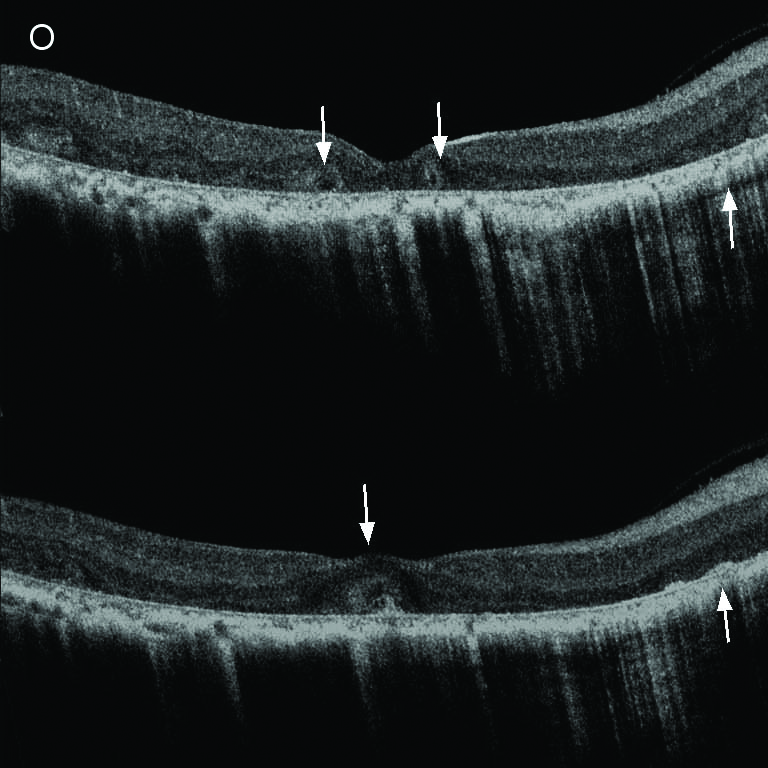
|
EDI
|
Choroidal layers clearly delineated; no pronounced atrophy
|
Focal or extensive loss of choriocapillaris; enhanced and more anterior visibility of larger outer choroidal vessels.
|
Severe atrophy and thinning of choroid; with the choroidal vessels virtually absent.
|
Table 3 Characterization of Multiple Imaging Modalities in Stages
| Imaging Modality | Stage1 | Stage 2 | Stage 3 |
| FAF | Normal AF or hyper/hypo-AF confined in the posterior pole | Confluent absent-AF patches and hypo-AF extend beyond the posterior pole | Prominent absent-AF in the posterior pole |
| SD-OCT | Drusen-like structure, loss of IZ and localized disruption of EZ in macula | Extensive loss of the outer retina and RPE, with focal residual RPE and/or EZ in macula | Complete loss of RPE and severe dystrophy of the macula |
| EDI | Choroidal layers clearly delineated; no pronounced atrophy | Focal or extensive loss of choriocapillaris; enhanced and more anterior visibility of larger outer choroidal vessels | Severe atrophy and thinning of choroid; with the choroidal vessels virtually absent |
FAF: fundus autofluorescence; SD-OCT: spectral-domain optical coherence tomography; EDI: enhanced depth imaging; AF: autofluorescence; IZ: interdigitation zone; EZ: ellipsoid zone; RPE: retinal pigment epithelium
Note:
- In early stage/young BCD patients, crystalline deposits may not be obvious and drusen-like structure and associated degeneration may be observed.
- In late stage BCD, crystalline deposits may diminish.
- Researchers suggested using infrared imaging to enhance retinal crystals in BCD. 4 5 In particular, hyperreflective appearance on near-infrared reflectance (NIR) imaging has been reported to yield 100% sensitivity and 100% specificity in diagnosing BCD among patients with chorioretinal dystrophy accompanied by crystalline-like deposits. 6
Misdiagnosis:
Misdiagnosis has been reported to be a serious issue related to BCD diagnosis. Many BCD cases could have been missed and misdiagnosed. See BCD Misdiagnosis for more detailed discussion, including the names of some diseases which BCD is sometimes misdiagnosed of.
Imaging Techniques
Multiple imaging techniques can be used in diagnosing BCD, e.g., autofluorescence (AF), fundus autofluorescence (FAF), spectral-domain optical coherence tomography (SD-OCT) and infared light.
Early-stage BCD:
In early stage/young BCD patients, crystalline deposits may not be obvious and drusen-like structure and associated degeneration may be observed.
Distinguish late-stage BCD from late-stage Choroideremia and other late-stage RP:
Because crystalline deposits in BCD may diminish in the advanced stage of the disease, late-stage BCD may be indistinguishable from other forms of retinal degeneration that fall under the category of retinitis pigmentosa (RP). Researchers suggested using infrared imaging to enhance retinal crystals in BCD (see endnotes 4 and 5).
Distinguish BCD from other Crystalline Retinopathies:
BCD is not the only eye disease that has crystalline deposits in the retina. There are other types of crystalline retinopathies. Please see relevant medical literature for discussions on and comparison of BCD and some other retinopathies. 7
Representative BCD photos taken by these imaging techniques can be found and are discussed in numerous BCD medical literature. (See BCD Literature).
Cornea
In some but not all BCD patients, crystalline deposits are also seen in the corneal limbus in addition to in the retina. Crystalline deposits in the cornea are usually seen on slit lamp examination but are not necessary for diagnosis. Spectral microscopy may be appropriate if corneal crystalline deposits are too subtle to detect on slit lamp examination.
Electrophysiology
Full-field electroretinogram (ffERG) can show varying degrees of rod and cone dysfunction, ranging from normal to reduced amplitudes of scotopic and photopic responses to undetectable responses. Note: The ffERG can remain normal even in later stages of the disease.
Multifocal electroretinogram (mfERG) may detect regional areas of abnormal retinal function when the ffERG is normal, particularly in those regional phenotypes that predominantly affect the posterior pole.
Genetic Testing:
Given the misdiagnosis and under-diagnosis issue (See BCD Misdiagnosis related to BCD, identification of biallelic mutations in CYP4V2 by molecular genetic testing can confirm the diagnosis if clinical features are inconclusive. To date, more than 100 mutations in the CYP4V2 gene has been identified among BCD patients. (See https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6679682/table/T0002/?report=objectonly 8 )
Molecular genetic testing approaches can include gene-targeted testing (single-gene testing or multigene panel). Depending on the phenotype, more comprehensive genomic testing (exome sequencing, exome array, genome sequencing) could be considered if CYP4V2 mutations are not found by targeted testing.
Note: Depending on the genetic testing approach and analysis used, a genetic testing may not identify all pathogenic mutations. In addition, pathogenic CYP4V2 mutations are not limited to those which have been reported in medical journals. More CYP4V2 mutations may be identified in the future.